Publications

Shafer et al. Gene family evolution underlies cell type diversification in the hypothalamus of teleosts. bioRxiv (2021)
Hundreds of cell types form the vertebrate brain, but it is largely unknown how similar these cellular repertoires are between or within species, or how cell type diversity evolves. To examine cell type diversity across and within species, we performed single-cell RNA sequencing of ~130,000 hypothalamic cells from zebrafish ( Danio rerio ) and surface- and cave-morphs of Mexican tetra ( Astyanax mexicanus ). We found that over 75% of cell types were shared between zebrafish and Mexican tetra, which last shared a common ancestor over 150 million years ago. Orthologous cell types displayed differential paralogue expression that was generated by sub-functionalization after genome duplication. Expression of terminal effector genes, such as neuropeptides, was more conserved than the expression of their associated transcriptional regulators. Species-specific cell types were enriched for the expression of species-specific genes, and characterized by the neo-functionalization of members of recently expanded or contracted gene families. Within species comparisons revealed differences in immune repertoires and transcriptional changes in neuropeptidergic cell types associated with genomic differences between surface- and cave-morphs. The single-cell atlases presented here are a powerful resource to explore hypothalamic cell types, and reveal how gene family evolution and the neo- and sub-functionalization of paralogs contribute to cellular diversity.
View Publication
Anderson et al. Simultaneous Identification of Brain Cell Type and Lineage via Single Cell RNA Sequencing. bioRxiv (2021)
Acquired mutations are sufficiently frequent such that the genome of a single cell offers a record of its history of cell divisions. Among more common somatic genomic alterations are loss of heterozygosity (LOH). Large LOH events are potentially detectable in single cell RNA sequencing (scRNA-seq) datasets as tracts of monoallelic expression for constitutionally heterozygous single nucleotide variants (SNVs) located among contiguous genes. We identified runs of monoallelic expression, consistent with LOH, uniquely distributed throughout the genome in single cell brain cortex transcriptomes of F1 hybrids involving different inbred mouse strains. We then phylogenetically reconstructed single cell lineages and simultaneously identified cell types by corresponding gene expression patterns. Our results are consistent with progenitor cells giving rise to multiple cortical cell types through stereotyped expansion and distinct waves of neurogenesis. Compared to engineered recording systems, LOH events accumulate throughout the genome and across the lifetime of an organism, affording tremendous capacity for encoding lineage information and increasing resolution for later cell divisions. This approach can conceivably be computationally incorporated into scRNA-seq analysis and may be useful for organisms where genetic engineering is prohibitive, such as humans.
View Publication
Lohoff et al. Highly multiplexed spatially resolved gene expression profiling of mouse organogenesis. bioRxiv (2020)
Transcriptional and epigenetic profiling of single-cells has advanced our knowledge of the molecular bases of gastrulation and early organogenesis. However, current approaches rely on dissociating cells from tissues, thereby losing the crucial spatial context that is necessary for understanding cell and tissue interactions during development. Here, we apply an image-based single-cell transcriptomics method, seqFISH, to simultaneously and precisely detect mRNA molecules for 387 selected target genes in 8-12 somite stage mouse embryo tissue sections. By integrating spatial context and highly multiplexed transcriptional measurements with two singlecell transcriptome atlases we accurately characterize cell types across the embryo and demonstrate how spatially-resolved expression of genes not profiled by seqFISH can be imputed. We use this high-resolution spatial map to characterize fundamental steps in the patterning of the midbrain-hindbrain boundary and the developing gut tube. Our spatial atlas uncovers axes of resolution that are not apparent from single-cell RNA sequencing data – for example, in the gut tube we observe early dorsal-ventral separation of esophageal and tracheal progenitor populations. In sum, by computationally integrating high-resolution spatially-resolved gene expression maps with single-cell genomics data, we provide a powerful new approach for studying how and when cell fate decisions are made during early mammalian development.
View Publication
Chow et al. Imaging cell lineage with a synthetic digital recording system. bioRxiv (2020)
Multicellular development depends on the differentiation of cells into specific fates with precise spatial organization. Lineage history plays a pivotal role in cell fate decisions, but is inaccessible in most contexts. Engineering cells to actively record lineage information in a format readable in situ would provide a spatially resolved view of lineage in diverse developmental processes. Here, we introduce a serine integrase-based recording system that allows in situ readout, and demonstrate its ability to reconstruct lineage relationships in cultured stem cells and flies. The system, termed intMEMOIR, employs an array of independent three-state genetic memory elements that can recombine stochastically and irreversibly, allowing up to 59,049 distinct digital states. intMEMOIR accurately reconstructed lineage trees in stem cells and enabled simultaneous analysis of single cell clonal history, spatial position, and gene expression in Drosophila brain sections. These results establish a foundation for microscopy-readable clonal analysis and recording in diverse systems.
View Publication
Takei et al. Integrated spatial genomics reveals global architecture of single nuclei. Nature (2021)
Identifying the relationships between chromosome structures, nuclear bodies, chromatin states and gene expression is an overarching goal of nuclear-organization studies1–4. Because individual cells appear to be highly variable at all these levels5, it is essential to map different modalities in the same cells. Here we report the imaging of 3,660 chromosomal loci in single mouse embryonic stem (ES) cells using DNA seqFISH+, along with 17 chromatin marks and subnuclear structures by sequential immunofluorescence and the expression profile of 70 RNAs. Many loci were invariably associated with immunofluorescence marks in single mouse ES cells. These loci form ‘fixed points’ in the nuclear organizations of single cells and often appear on the surfaces of nuclear bodies and zones defined by combinatorial chromatin marks. Furthermore, highly expressed genes appear to be pre-positioned to active nuclear zones, independent of bursting dynamics in single cells. Our analysis also uncovered several distinct mouse ES cell subpopulations with characteristic combinatorial chromatin states. Using clonal analysis, we show that the global levels of some chromatin marks, such as H3 trimethylation at lysine 27 (H3K27me3) and macroH2A1 (mH2A1), are heritable over at least 3–4 generations, whereas other marks fluctuate on a faster time scale. This seqFISH+-based spatial multimodal approach can be used to explore nuclear organization and cell states in diverse biological systems.
View Publication
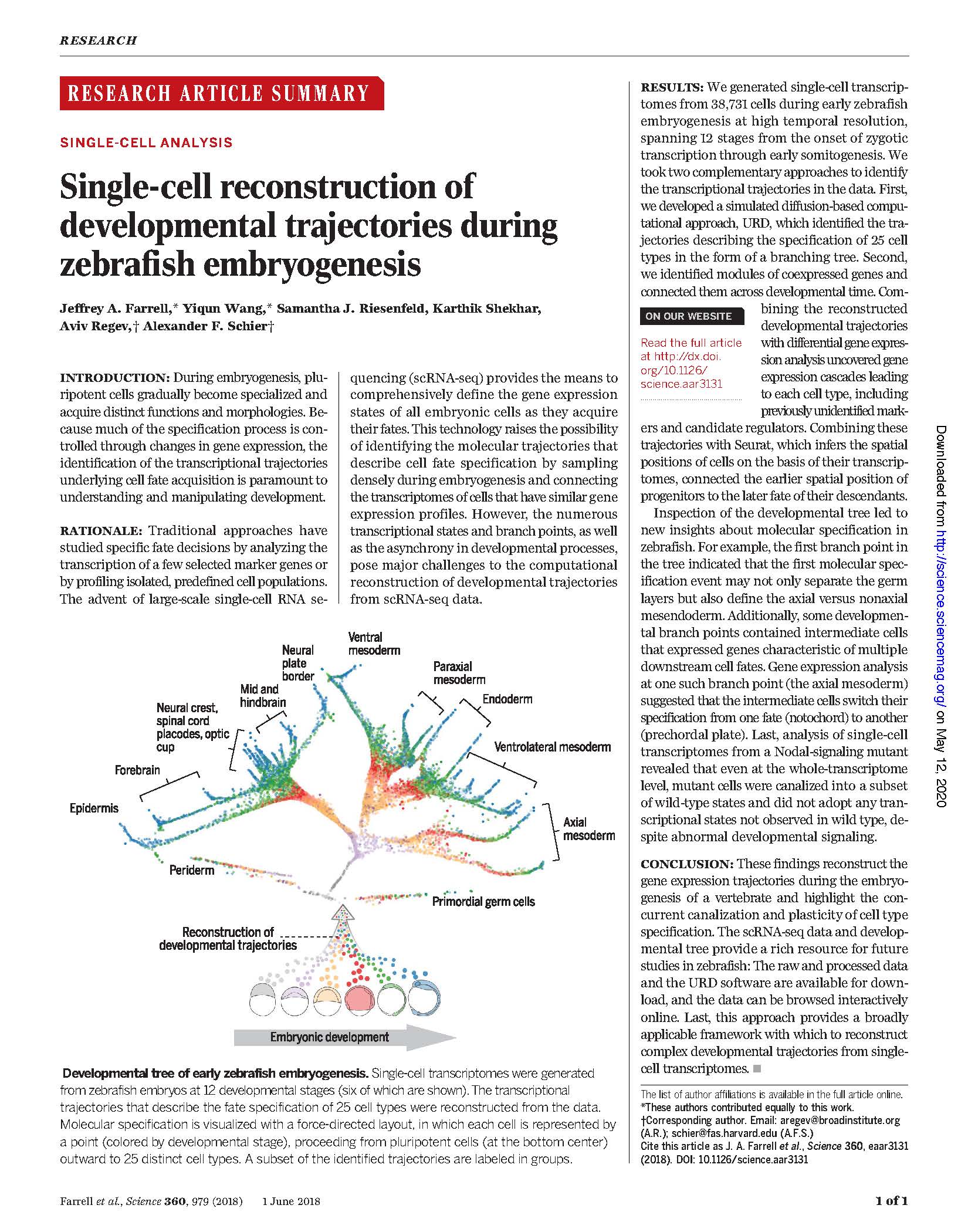
Raj et al. Emergence of neuronal diversity during vertebrate brain development. Neuron (2020).
Neurogenesis in the vertebrate brain comprises many steps ranging from the proliferation of progenitors to the differentiation and maturation of neurons. Although these processes are highly regulated, the landscape of transcriptional changes and progenitor identities underlying brain development are poorly characterized. Here, we describe the first developmental single-cell RNA-seq catalog of more than 200,000 zebrafish brain cells encompassing 12 stages from 12 hours post-fertilization to 15 days post-fertilization. We characterize known and novel gene markers for more than 800 clusters across these timepoints. Our results capture the temporal dynamics of multiple neurogenic waves from embryo to larva that expand neuronal diversity from ∼20 cell types at 12 hpf to ∼100 cell types at 15 dpf. We find that most embryonic neural progenitor states are transient and transcriptionally distinct from long-lasting neural progenitors of post-embryonic stages. Furthermore, we reconstruct cell specification trajectories for the retina and hypothalamus, and identify gene expression cascades and novel markers. Our analysis reveal that late-stage retinal neural progenitors transcriptionally overlap cell states observed in the embryo, while hypothalamic neural progenitors become progressively distinct with developmental time. These data provide the first comprehensive single-cell transcriptomic time course for vertebrate brain development and suggest distinct neurogenic regulatory paradigms between different stages and tissues.
View Publication
Domcke et al. A human cell atlas of fetal chromatin accessibility. Science 2020
In recent years, the single-cell genomics field has made incredible progress toward disentangling the cellular heterogeneity of human tissues. However, the overwhelming majority of effort has been focused on singlecell gene expression rather than the chromatin landscape that shapes and is shaped by gene expression. Toward advancing our understanding of the regulatory programs that underlie human cell types, we set out to generate singlecell atlases of both chromatin accessibility (this study) and gene expression (Cao et al., this issue) from a broad range of human fetal tissues.
View Publication
Cao J et al. A human cell atlas of fetal gene expression. Science (2020)
A reference atlas of human cell types is a major goal for the field. Here,we set out to generate single-cell atlases of both gene expression (this study) and chromatin accessibility (Domcke et al., this issue) using diverse human tissues obtained during midgestation.
View Publication
Cao et al. Sci-fate characterizes the dynamics of gene expression in single cells. Nature Biotechnology (2020)
Gene expression programs change over time, differentiation and development, and in response to stimuli. However, nearly all techniques for profiling gene expression in single cells do not directly capture transcriptional dynamics. In the present study, we present a method for combined single-cell combinatorial indexing and messenger RNA labeling (sci-fate), which uses combinatorial cell indexing and 4-thiouridine labeling of newly synthesized mRNA to concurrently profile the whole and newly synthesized transcriptome in each of many single cells. We used sci-fate to study the cortisol response in >6,000 single cultured cells. From these data, we quantified the dynamics of the cell cycle and glucocorticoid receptor activation, and explored their intersection. Finally, we developed software to infer and analyze cell-state transitions. We anticipate that sci-fate will be broadly applicable to quantitatively characterize transcriptional dynamics in diverse systems.
View Publication
Schier. Single-cell biology: beyond the sum of its parts. Nature Methods (2020)
The field of single-cell RNA sequencing (scRNA-seq) has been paired with genomics, epigenomics, spatial omics, proteomics and imaging to achieve multimodal measurements of individual cellular phenotypes and genotypes. In its purest form, single-cell multimodal omics involves the simultaneous detection of multiple traits in the same cell. More broadly, multimodal omics also encompasses comparative pairing and computational integration of measurements made across multiple distinct cells to reconstruct phenotypes. Here I highlight some of the biological insights gained from multimodal studies and discuss the challenges and opportunities in this emerging field.
View Publication
Askary et al. In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nature Biotechnology (2020).
Molecular barcoding technologies that uniquely identify single cells are hampered by limitations in barcode measurement.Readout by sequencing does not preserve the spatial organization of cells in tissues, whereas imaging methods preserve spatial structure but are less sensitive to barcode sequence. Here we introduce a system for image-based readout of short (20-basepair) DNA barcodes. In this system, called Zombie, phage RNA polymerases transcribe engineered barcodes in fixed cells. Theresulting RNA is subsequently detected by fluorescent in situ hybridization. Using competing match and mismatch probes,Zombie can accurately discriminate single-nucleotide differences in the barcodes. This method allows in situ readout of densecombinatorial barcode libraries and single-base mutations produced by CRISPR base editors without requiring barcode expressionin live cells. Zombie functions across diverse contexts, including cell culture, chick embryos and adult mouse brain tissue. The ability to sensitively read out compact and diverse DNA barcodes by imaging will facilitate a broad range of barcoding and genomic recording strategies.
View Publication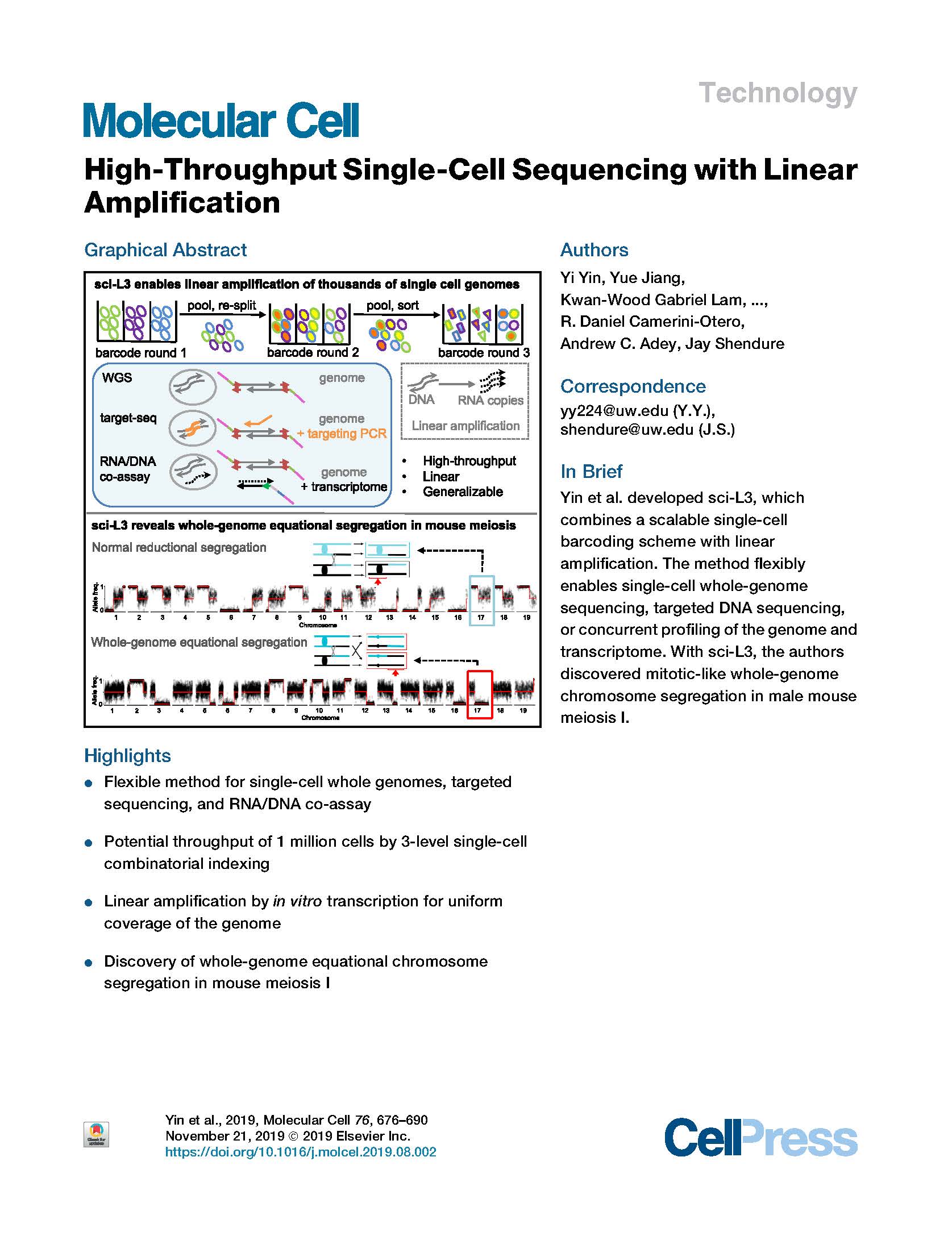
Yin et al. High-throughput single cell sequencing with linear amplification. Molecular Cell (2019).
Conventional methods for single-cell genome sequencing are limited with respect to uniformity and throughput. Here, we describe sci-L3, a single-cell sequencing method that combines combinatorial indexing (sci-) and linear (L) amplification. The sci-L3 method adopts a 3-level (3) indexing scheme that minimizes amplification biases while enabling exponential gains in throughput. We demonstrate the generalizability of sci-L3 with proof-of-concept demonstrations of single-cell whole-genome sequencing (sci-L3-WGS), targeted sequencing (sci-L3-targetseq), and a co-assay of the genome and transcriptome (sci-L3-RNA/DNA). We apply sci-L3-WGS to profile the genomes of >10,000 sperm and sperm precursors from F1 hybrid mice, mapping 86,786 crossovers and characterizing rare chromosome mis-segregation events in meiosis, including instances of whole-genome equational chromosome segregation. We anticipate that sci-L3 assays can be applied to fully characterize recombination landscapes, to couple CRISPR perturbations and measurements of genome stability, and to other goals requiring high-throughput, high-coverage single-cell sequencing.
View Publication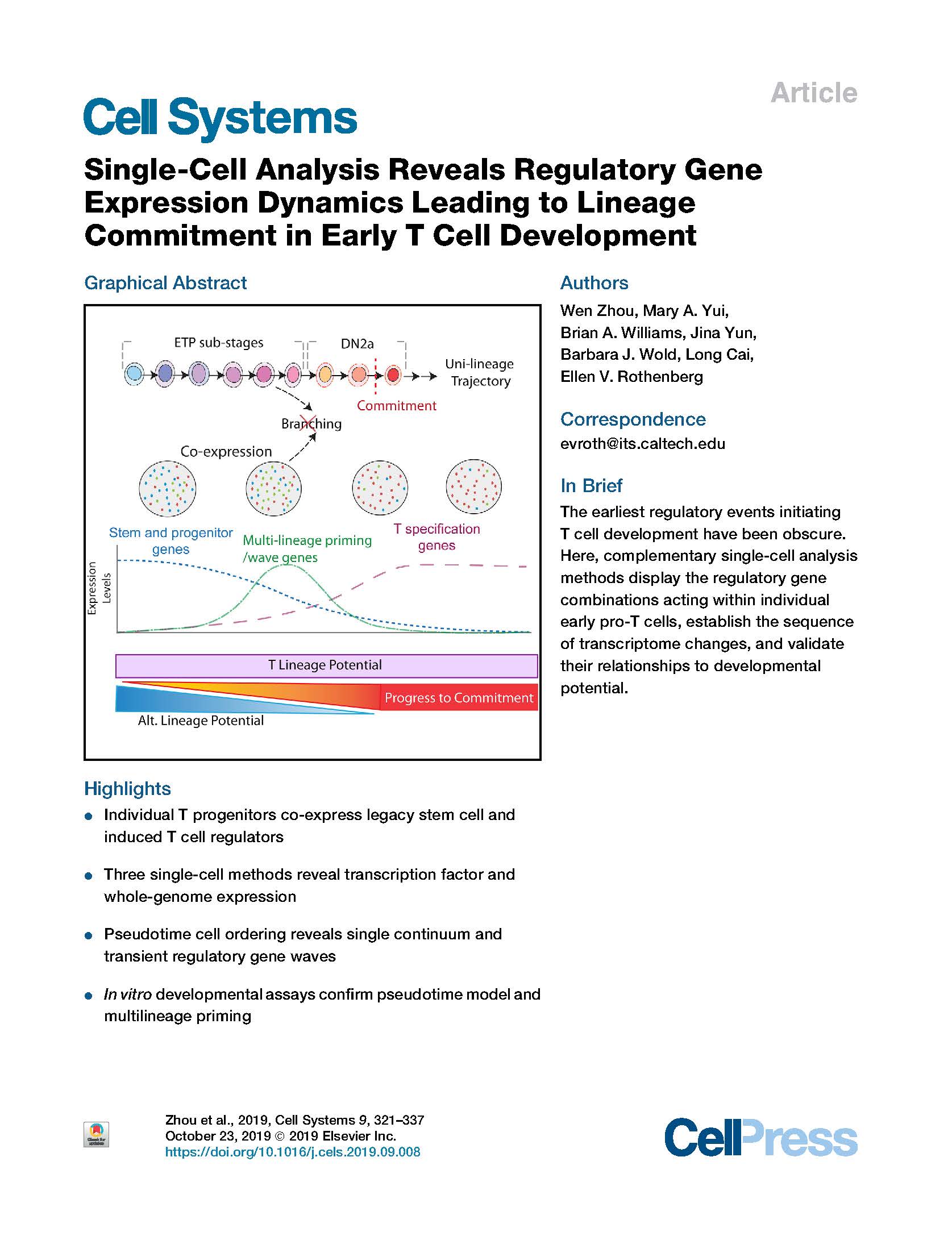
Zhou et al. Single-Cell Analysis Reveals Regulatory Gene Expression Dynamics Leading to Lineage Commitment in Early T Cell Development. Cell Systems (2019).
Intrathymic T cell development converts multipotent precursors to committed pro-T cells, silencing progenitor genes while inducing T cell genes, but the underlying steps have remained obscure. Single-cell profiling was used to define the order of regulatory changes, employing single-cell RNA sequencing (scRNA-seq) for full-transcriptome analysis, plus sequential multiplexed single-molecule fluorescent in situ hybridization (seqFISH) to quantitate functionally important transcripts in intrathymic precursors. Single-cell cloning verified high T cell precursor frequency among the immunophenotypically defined “early T cell precursor” (ETP) population; a discrete committed granulocyte precursor subset was also distinguished. We established regulatory phenotypes of sequential ETP subsets, confirmed initial co-expression of progenitor with T cell specification genes, defined stage-specific relationships between cell cycle and differentiation, and generated a pseudotime model from ETP to T lineage commitment, supported by RNA velocity and transcription factor perturbations. This model was validated by developmental kinetics of ETP subsets at population and clonal levels. The results imply that multilineage priming is integral to T cell specification.
View Publication
Kim et al. Multimodal Analysis of Cell Types in a Hypothalamic Node Controlling Social Behavior. Cell (2019).
The ventrolateral subdivision of the ventromedial hypothalamus (VMHvl) contains ∼4,000 neurons that project to multiple targets and control innate social behaviors including aggression and mounting. However, the number of cell types in VMHvl and their relationship to connectivity and behavioral function are unknown. We performed single-cell RNA sequencing using two independent platforms—SMART-seq (∼4,500 neurons) and 10x (∼78,000 neurons)—and investigated correspondence between transcriptomic identity and axonal projections or behavioral activation, respectively. Canonical correlation analysis (CCA) identified 17 transcriptomic types (T-types), including several sexually dimorphic clusters, the majority of which were validated by seqFISH. Immediate early gene analysis identified T-types exhibiting preferential responses to intruder males versus females but only rare examples of behavior-specific activation. Unexpectedly, many VMHvl T-types comprise a mixed population of neurons with different projection target preferences. Overall our analysis revealed that, surprisingly, few VMHvl T-types exhibit a clear correspondence with behavior-specific activation and connectivity.
View Publication
Pliner et al. Supervised classification enables rapid annotation of cell atlases. Nature Methods (2019).
Single-cell molecular profiling technologies are gaining rapid traction, but the manual process by which resulting cell types are typically annotated is labor intensive and rate-limiting. We describe Garnett, a tool for rapidly annotating cell types in single-cell transcriptional profiling and single-cell chromatin accessibility datasets, based on an interpretable, hierarchical markup language of cell type-specific genes. Garnett successfully classifies cell types in tissue and whole organism datasets, as well as across species.
View Publication
McFaline-Figueroa et al. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nature Genetics (2019).
Integrating single-cell trajectory analysis with pooled genetic screening could reveal the genetic architecture that guides cellular decisions in development and disease. We applied this paradigm to probe the genetic circuitry that controls epithelial-to-mesenchymal transition (EMT). We used single-cell RNA sequencing to profile epithelial cells undergoing a spontaneous spatially determined EMT in the presence or absence of transforming growth factor-β. Pseudospatial trajectory analysis identified continuous waves of gene regulation as opposed to discrete ‘partial’ stages of EMT. KRAS was connected to the exit from the epithelial state and the acquisition of a fully mesenchymal phenotype. A pooled single-cell CRISPR-Cas9 screen identified EMT-associated receptors and transcription factors, including regulators of KRAS, whose loss impeded progress along the EMT. Inhibiting the KRAS effector MEK and its upstream activators EGFR and MET demonstrates that interruption of key signaling events reveals regulatory ‘checkpoints’ in the EMT continuum that mimic discrete stages, and reconciles opposing views of the program that controls EMT.
View Publication
Packer et al. A lineage-resolved molecular atlas of C. elegans embryogenesis at single cell resolution. Science (2019).
During development, a single-cell zygote undergoes repeated cell divisions to produce an embryo that contains many distinct cell types. This sequence of cell divisions is called the organism’s cell lineage. Each cell in the lineage expresses a different set of genes in various quantities (the cell’s transcriptome), thus directing cells to differentiate into specific cell types over time. Identifying these transcriptome changes and understanding how they regulate cell type specification are fundamental challenges in biology. Advances in methods that assay the mRNA content of single cells [e.g., single-cell RNA sequencing (sc-RNA-seq)] have made it possible to directly measure these gene expression patterns for each cell at a whole-organism scale.
View Publication
Chen et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Research (2019).
Non-homologous end-joining (NHEJ) plays an important role in double-strand break (DSB) repair of DNA. Recent studies have shown that the error patterns of NHEJ are strongly biased by sequence context, but these studies were based on relatively few templates. To investigate this more thoroughly, we systematically profiled ∼1.16 million independent mutational events resulting from CRISPR/Cas9-mediated cleavage and NHEJ-mediated DSB repair of 6872 synthetic target sequences, introduced into a human cell line via lentiviral infection. We find that: (i) insertions are dominated by 1 bp events templated by sequence immediately upstream of the cleavage site, (ii) deletions are predominantly associated with microhomology and (iii) targets exhibit variable but reproducible diversity with respect to the number and relative frequency of the mutational outcomes to which they give rise. From these data, we trained a model that uses local sequence context to predict the distribution of mutational outcomes. Exploiting the bias of NHEJ outcomes towards microhomology mediated events, we demonstrate the programming of deletion patterns by introducing microhomology to specific locations in the vicinity of the DSB site. We anticipate that our results will inform investigations of DSB repair mechanisms as well as the design of CRISPR/Cas9 experiments for diverse applications including genome-wide screens, gene therapy, lineage tracing and molecular recording.
View Publication
Sánchez-Guardado and Lois. Lineage does not regulate the sensory synaptic input of projection neurons in the mouse olfactory bulb. eLife (2019).
Lineage regulates the synaptic connections between neurons in some regions of the invertebrate nervous system. In mammals, recent experiments suggest that cell lineage determines the connectivity of pyramidal neurons in the neocortex, but the functional relevance of this phenomenon and whether it occurs in other neuronal types remains controversial. We investigated whether lineage plays a role in the connectivity of mitral and tufted cells, the projection neurons in the mouse olfactory bulb. We used transgenic mice to sparsely label neuronal progenitors and observed that clonally related neurons receive synaptic input from olfactory sensory neurons expressing different olfactory receptors. These results indicate that lineage does not determine the connectivity between olfactory sensory neurons and olfactory bulb projection neurons.
View Publication
Dries et al. Giotto, a pipeline for integrative analysis and visualization of single-cell spatial transcriptomic data. bioRxiv (2019).
The rapid development of novel spatial transcriptomics technologies has provided new opportunities to investigate the interactions between cells and their native microenvironment. However, effective use of such technologies requires the development of innovative computational algorithms and pipelines. Here we present Giotto, a comprehensive, flexible, robust, and open-source pipeline for spatial transcriptomic data analysis and visualization. The data analysis module implements a wide range of algorithms ranging from basic tasks such as data pre-processing to innovative approaches for cell-cell interaction characterization. The data visualization module provides a user-friendly workspace that allows users to interactively visualize, explore and compare multiple layers of information. These two modules can be used iteratively for refined analysis and hypothesis development. We illustrate the functionalities of Giotto by using the recently published seqFISH+ dataset for mouse brain. Our analysis highlights the utility of Giotto for characterizing tissue spatial organization as well as for the interactive exploration of multi- layer information in spatial transcriptomic and imaging data. We find that single-cell resolution spatial information is essential for the investigation of ligand-receptor mediated cell-cell interactions. Giotto is generally applicable and can be easily integrated with external software packages for multi-omic data integration.
View Publication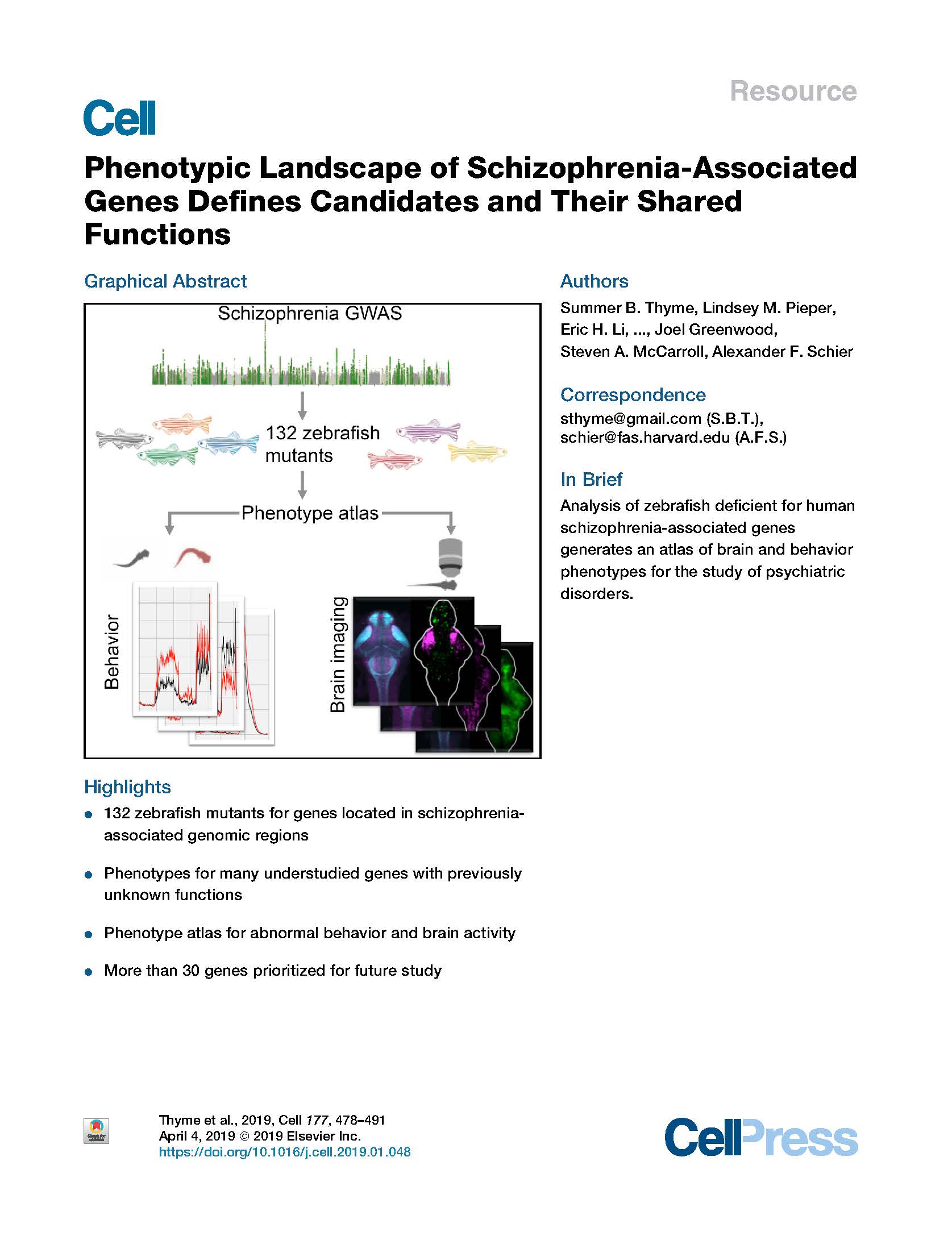
Thyme et al. Phenotypic Landscape of Schizophrenia-Associated Genes Defines Candidates and Their Shared Functions. Cell (2019).
Genomic studies have identified hundreds of candidate genes near loci associated with risk for schizophrenia. To define candidates and their functions, we mutated zebrafish orthologs of 132 human schizophrenia-associated genes. We created a phenotype atlas consisting of whole-brain activity maps, brain structural differences, and profiles of behavioral abnormalities. Phenotypes were diverse but specific, including altered forebrain development and decreased prepulse inhibition. Exploration of these datasets identified promising candidates in more than 10 gene-rich regions, including the magnesium transporter cnnm2 and the translational repressor gigyf2, and revealed shared anatomical sites of activity differences, including the pallium, hypothalamus, and tectum. Single-cell RNA sequencing uncovered an essential role for the understudied transcription factor znf536 in the development of forebrain neurons implicated in social behavior and stress. This phenotypic landscape of schizophrenia-associated genes prioritizes more than 30 candidates for further study and provides hypotheses to bridge the divide between genetic association and biological mechanism.
View Publication
Eng et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature (2019).
Imaging the transcriptome in situ with high accuracy has been a major challenge in single-cell biology, which is particularly hindered by the limits of optical resolution and the density of transcripts in single cells1–5. Here we demonstrate an evolution of sequential fluorescence in situ hybridization (seqFISH+). We show that seqFISH+ can image mRNAs for 10,000 genes in single cells—with high accuracy and sub-diffraction-limit resolution—in the cortex, subventricular zone and olfactory bulb of mouse brain, using a standard confocal microscope. The transcriptome-level profiling of seqFISH+ allows unbiased identification of cell classes and their spatial organization in tissues. In addition, seqFISH+ reveals subcellular mRNA localization patterns in cells and ligand–receptor pairs across neighbouring cells. This technology demonstrates the ability to generate spatial cell atlases and to perform discoverydriven studies of biological processes in situ.
View Publication
Cao, Spielmann et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature (2019).
Mammalian organogenesis is a remarkable process. Within a short timeframe, the cells of the three germ layers transform into an embryo that includes most of the major internal and external organs. Here we investigate the transcriptional dynamics of mouse organogenesis at single-cell resolution. Using single-cell combinatorial indexing, we profiled the transcriptomes of around 2 million cells derived from 61 embryos staged between 9.5 and 13.5 days of gestation, in a single experiment. The resulting ‘mouse organogenesis cell atlas’ (MOCA) provides a global view of developmental processes during this critical window. We use Monocle 3 to identify hundreds of cell types and 56 trajectories, many of which are detected only because of the depth of cellular coverage, and collectively define thousands of corresponding marker genes. We explore the dynamics of gene expression within cell types and trajectories over time, including focused analyses of the apical ectodermal ridge, limb mesenchyme and skeletal muscle.
View Publication
Raj et al. Large-scale reconstruction of cell lineages using single-cell readout of transcriptomes and CRISPR-Cas9 barcodes by scGESTALT. Nature Protocols (2018).
Lineage relationships among the large number of heterogeneous cell types generated during development are difficult to reconstruct in a high-throughput manner. We recently established a method, scGESTALT, that combines cumulative editing of a lineage barcode array by CRISPR–Cas9 with large-scale transcriptional profiling using droplet-based singlecell RNA sequencing (scRNA-seq). The technique generates edits in the barcode array over multiple timepoints using Cas9 and pools of single-guide RNAs (sgRNAs) introduced during early and late zebrafish embryonic development, which distinguishes it from similar Cas9 lineage-tracing methods. The recorded lineages are captured, along with thousands of cellular transcriptomes, to build lineage trees with hundreds of branches representing relationships among profiled cell types. Here, we provide details for (i) generating transgenic zebrafish; (ii) performing multi-timepoint barcode editing; (iii) building scRNA-seq libraries from brain tissue; and (iv) concurrently amplifying lineage barcodes from captured single cells. Generating transgenic lines takes 6 months, and performing barcode editing and generating single-cell libraries involve 7 d of hands-on time. scGESTALT provides a scalable platform to map lineage relationships between cell types in any system that permits genome editing during development, regeneration, or disease.
View Publication
Cao et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science (2018).
Although we can increasingly measure transcription, chromatin, methylation, and other aspects of molecular biology at single-cell resolution, most assays survey only one aspect of cellular biology. Here we describe sci-CAR, a combinatorial indexing–based coassay that jointly profiles chromatin accessibility and mRNA (CAR) in each of thousands of single cells. As a proof of concept, we apply sci-CAR to 4825 cells, including a time series of dexamethasone treatment, as well as to 11,296 cells from the adult mouse kidney. With the resulting data, we compare the pseudotemporal dynamics of chromatin accessibility and gene expression, reconstruct the chromatin accessibility profiles of cell types defined by RNA profiles, and link cis-regulatory sites to their target genes on the basis of the covariance of chromatin accessibility and transcription across large numbers of single cells.
View Publication
Hart et al. Activating PAX gene family paralogs to complement PAX5 leukemia driver mutations. PLOS Genetics (2018)
PAX5, one of nine members of the mammalian paired box (PAX) family of transcription factors, plays an important role in B cell development. Approximately one-third of individuals with pre-B acute lymphoblastic leukemia (ALL) acquire heterozygous inactivating mutations of PAX5 in malignant cells, and heterozygous germline loss-of-function PAX5 mutations cause autosomal dominant predisposition to ALL. At least in mice, Pax5 is required for pre- B cell maturation, and leukemic remission occurs when Pax5 expression is restored in a Pax5-deficient mouse model of ALL. Together, these observations indicate that PAX5 deficiency reversibly drives leukemogenesis. PAX5 and its two most closely related paralogs, PAX2 and PAX8, which are not mutated in ALL, exhibit overlapping expression and function redundantly during embryonic development. However, PAX5 alone is expressed in lymphocytes, while PAX2 and PAX8 are predominantly specific to kidney and thyroid, respectively. We show that forced expression of PAX2 or PAX8 complements PAX5 loss-of-function mutation in ALL cells as determined by modulation of PAX5 target genes, restoration of immunophenotypic and morphological differentiation, and, ultimately, reduction of replicative potential. Activation of PAX5 paralogs, PAX2 or PAX8, ordinarily silenced in lymphocytes, may therefore represent a novel approach for treating PAX5-deficient ALL. In pursuit of this strategy, we took advantage of the fact that, in kidney, PAX2 is upregulated by extracellular hyperosmolarity. We found that hyperosmolarity, at potentially clinically achievable levels, transcriptionally activates endogenous PAX2 in ALL cells via a mechanism dependent on NFAT5, a transcription factor coordinating response to hyperosmolarity. We also found that hyperosmolarity upregulates residual wild type PAX5 expression in ALL cells and modulates gene expression, including in PAX5-mutant primary ALL cells. These findings specifically demonstrate that osmosensing pathways may represent a new therapeutic target for ALL and more broadly point toward the possibility of using gene paralogs to rescue mutations driving cancer and other diseases.
View Publication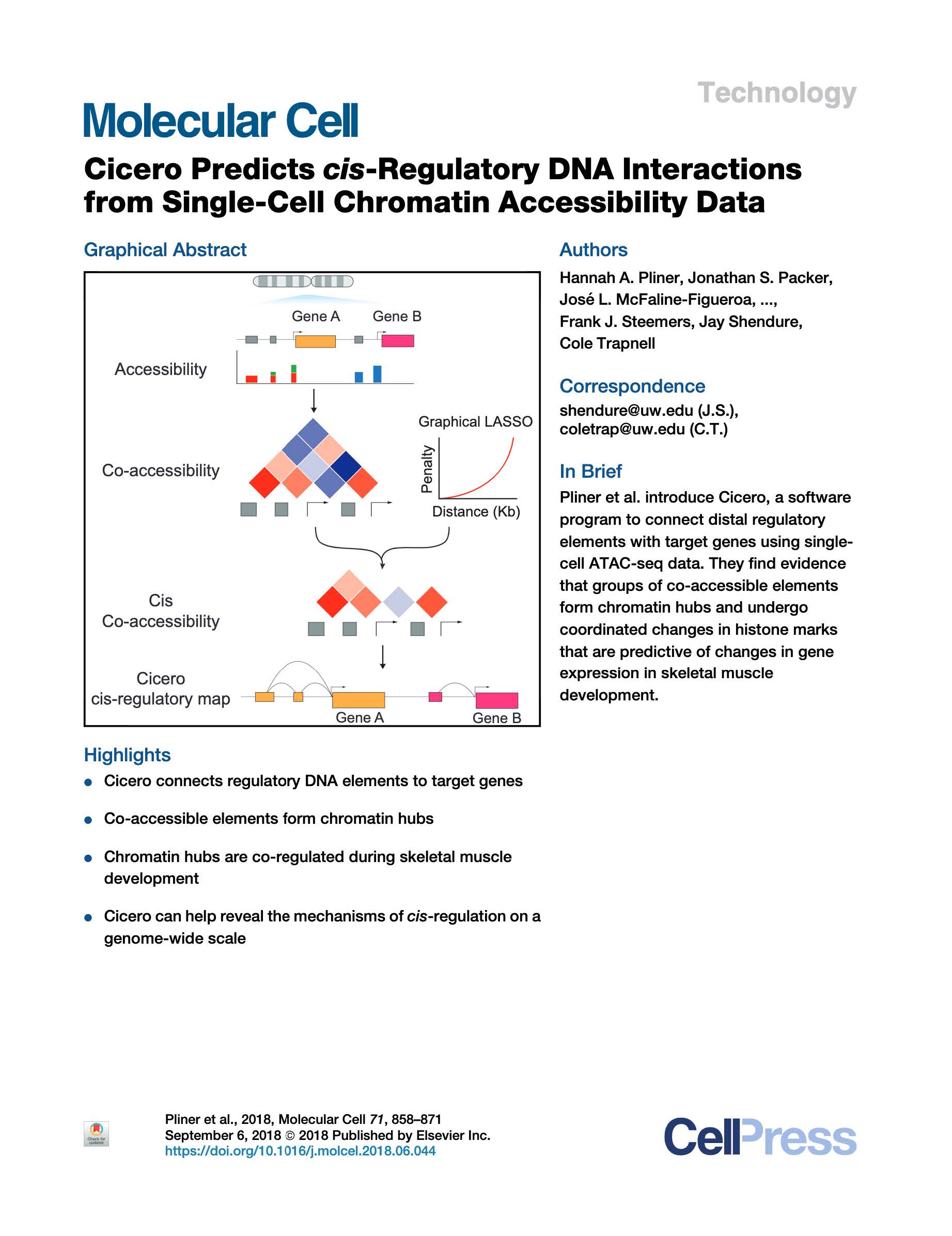
Pliner et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Molecular Cell (2018).
Linking regulatory DNA elements to their target genes, which may be located hundreds of kilobases away, remains challenging. Here, we introduce Cicero, an algorithm that identifies co-accessible pairs of DNA elements using single-cell chromatin accessibility data and so connects regulatory elements to their putative target genes. We apply Cicero to investigate how dynamically accessible elements orchestrate gene regulation in differentiating myoblasts. Groups of Cicero-linked regulatory elements meet criteria of "chromatin hubs"-they are enriched for physical proximity, interact with a common set of transcription factors, and undergo coordinated changes in histone marks that are predictive of changes in gene expression. Pseudotemporal analysis revealed that most DNA elements remain in chromatin hubs throughout differentiation. A subset of elements bound by MYOD1 in myoblasts exhibit early opening in a PBX1- and MEIS1 -dependent manner. Our strategy can be applied to dissect the architecture, sequence determinants, and mechanisms of cis-regulation on a genome-wide scale.
View Publication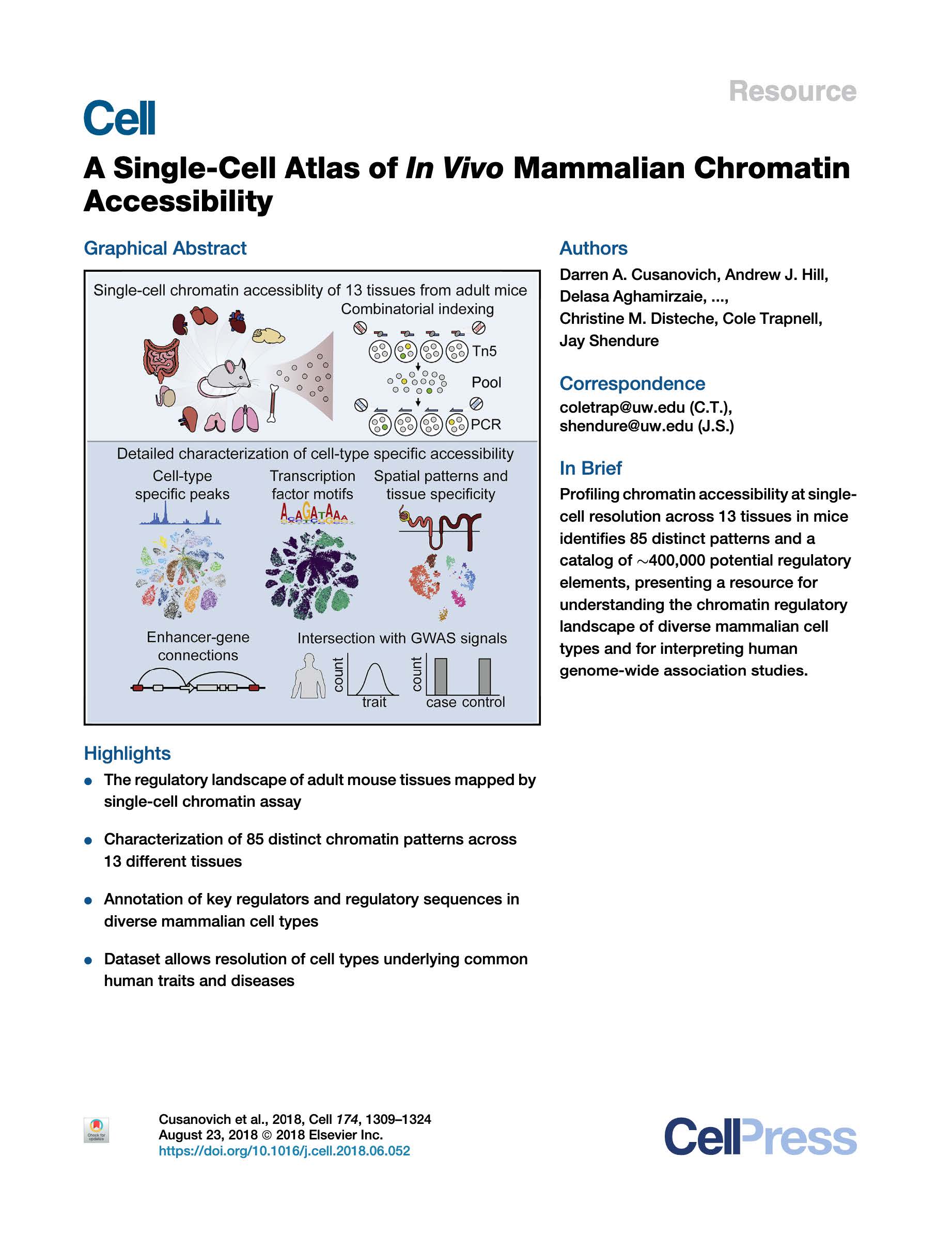
Cusanovich, Hill et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell (2018).
We applied a combinatorial indexing assay, sciATAC-seq, to profile genome-wide chromatin accessibility in ~100,000 single cells from 13 adult mouse tissues. We identify 85 distinct patterns of chromatin accessibility, most of which can be assigned to cell types, and ~400,000 differentially accessible elements. We use these data to link regulatory elements to their target genes, to define the transcription factor grammar specifying each cell type, and to discover in vivo correlates of heterogeneity in accessibility within cell types. We develop a technique for mapping single cell gene expression data to single-cell chromatin accessibility data, facilitating the comparison of atlases. By intersecting mouse chromatin accessibility with human genome-wide association summary statistics, we identify cell-type-specific enrichments of the heritability signal for hundreds of complex traits. These data define the in vivo landscape of the regulatory genome for common mammalian cell types at single-cell resolution.
View Publication
Shah et al. Dynamics and Spatial Genomics of the Nascent Transcriptome by Intron seqFISH. Cell (2018).
Visualization of the transcriptome and the nuclear organization in situ has been challenging for single-cell analysis. Here, we demonstrate a multiplexed single-molecule in situ method, intron seqFISH, that allows imaging of 10,421 genes at their nascent transcription active sites in single cells, followed by mRNA and lncRNA seqFISH and immunofluorescence. This nascent transcriptome-profiling method can identify different cell types and states with mouse embryonic stem cells and fibroblasts. The nascent sites of RNA synthesis tend to be localized on the surfaces of chromosome territories, and their organization in individual cells is highly variable. Surprisingly, the global nascent transcription oscillated asynchronously in individual cells with a period of 2 hr in mouse embryonic stem cells, as well as in fibroblasts. Together, spatial genomics of the nascent transcriptome by intron seqFISH reveals nuclear organizational principles and fast dynamics in single cells that are otherwise obscured.
View Publication
McKenna et al. FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biology (2018).
Genome-wide knockout studies, noncoding deletion scans, and other large-scale studies require a simple and lightweight framework that can quickly discover and score thousands of candidate CRISPR guides targeting an arbitrary DNA sequence. While several CRISPR web applications exist, there is a need for a highthroughput tool to rapidly discover and process hundreds of thousands of CRISPR targets. Here, we introduce FlashFry, a fast and flexible command-line tool for characterizing large numbers of CRISPR target sequences. With FlashFry, users can specify an unconstrained number of mismatches to putative offtargets, richly annotate discovered sites, and tag potential guides with commonly used on-target and off-target scoring metrics. FlashFry runs at speeds comparable to commonly used genome-wide sequence aligners, and output is provided as an easy-to-manipulate text file. FlashFry is a fast and convenient command-line tool to discover and score CRISPR targets within large DNA sequences..
View Publication
Raj et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nature Biotechnology (2018).
The lineage relationships among the hundreds of cell types generated during development are difficult to reconstruct. A recent method, GESTALT, used CRISPR-Cas9 barcode editing for large-scale lineage tracing, but was restricted to early development and did not identify cell types. Here we present scGESTALT, which combines the lineage recording capabilities of GESTALT with cell-type identification by single-cell RNA sequencing. The method relies on an inducible system that enables barcodes to be edited at multiple time points, capturing lineage information from later stages of development. Sequencing of ∼60,000 transcriptomes from the juvenile zebrafish brain identified >100 cell types and marker genes. Using these data, we generate lineage trees with hundreds of branches that help uncover restrictions at the level of cell types, brain regions, and gene expression cascades during differentiation. scGESTALT can be applied to other multicellular organisms to simultaneously characterize molecular identities and lineage histories of thousands of cells during development and disease.
View Publication